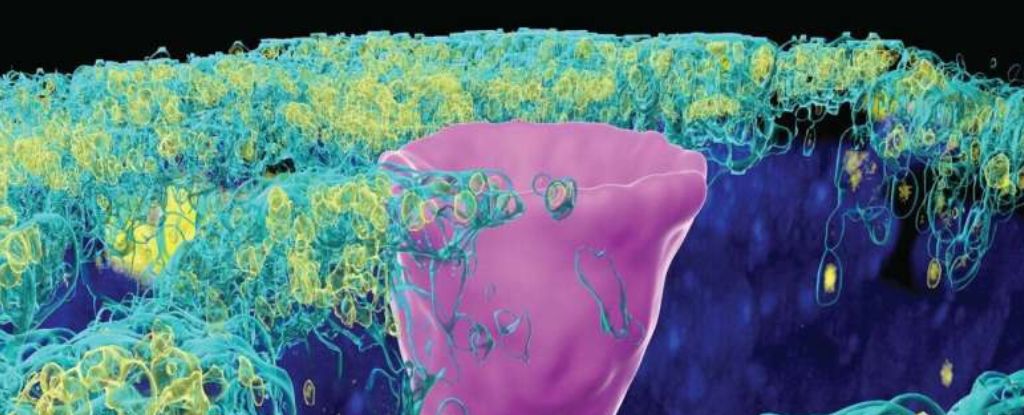
A type of cell once only thought to exist in the gills of freshwater fish and the skin of frogs, but recently found in humans lungs, has given scientists new insight into the underlying cause of cystic fibrosis (CF).
CF is a progressive, genetic disease that impacts the lungs and other organs, sometimes causing severe symptoms that can be life-threatening.
The disease is marked by the absence or mutation of a protein in the lungs called the cystic fibrosis transmembrane conductance regulator (CFTR).
This protein is known to regulate the balance of salt and water on the surface of the lung, but its structure and function are not yet fully understood by scientists.
New research, led by scientists at the University of Iowa, has now revealed an unexpected new function of the CFTR protein.
To date, most research on CFTR proteins has been conducted around airway secretory cells.
In these cells, CFTR channels, embedded in the surface of the cell, secrete chloride ions into the thin layer of liquid covering the airway surface.
Where salt goes, water follows, thereby re-hydrating this important liquid layer of the lungs. This layer of lubricating mucus traps irritants and pathogens, which can later be expelled by coughing.
Those with CF, however, have thick and sticky mucus, as opposed to thin and runny mucus, and this can plug up passageways in the lungs.
But here's the catch: CFTR proteins don't just show up in secretory cells. In 2018, researchers found a whole new type of cell hiding in the human lung, called ionoctyes, which are common in fish and frogs.
Pulmonary ionocytes make up only 1 percent of all the cells in the human lung, and yet they contain most of the CFTR channels in our bodies.
Culturing these cells in the lab, researchers have now caught CFTR proteins behaving very strangely.
Instead of secreting chloride, when embedded in ionocytes they seem to absorb the ions, thereby sucking moisture away from the liquid layer that lines the lungs.
In experiments, increasing the abundance of ionocytes increased liquid absorption, not liquid secretion.
What's more, disrupting the chloride ion channels in the ionoctyes impaired their absorption.
"These findings indicate that ionocytes mediate liquid absorption, and secretory cells mediate liquid secretion," write the University of Iowa team.
"Moreover, the divergent role of CFTR in ionocytes and secretory cells suggests that cystic fibrosis disrupts both liquid secretion and absorption."
If the researchers are right, then their results bring new understanding to the pathology of cystic fibrosis.
Previous studies that have stimulated CFTR channels have produced conflicting observations on how that increased activity impacts liquid transport in the lungs.
The new findings present a possible explanation for the confusion: the activity of CFTR proteins may depend on where they are located, and so far, we've missed their main residence.
Genetic therapies that target this cell type in the future could hold great promise for the treatment of CF.
"Much remains to be learned about how the individual transport processes in ionocytes are regulated," the researchers conclude.
"The observation that CFTR is critical for [chloride ion] absorption, in addition to [chloride ion] secretion, indicates that both processes are disrupted in CF."
The study was published in The Journal of Clinical Investigation.
"fish" - Google News
October 28, 2023 at 12:01AM
https://ift.tt/gUmE5Dc
Unexpected 'Fish' Cell Found in Human Lungs Could Be Key to Cystic Fibrosis - ScienceAlert
"fish" - Google News
https://ift.tt/1Im69aR
https://ift.tt/2Isaw86
Bagikan Berita Ini














0 Response to "Unexpected 'Fish' Cell Found in Human Lungs Could Be Key to Cystic Fibrosis - ScienceAlert"
Post a Comment